
Lim Heo and Michael Feig
Proteins (2019)
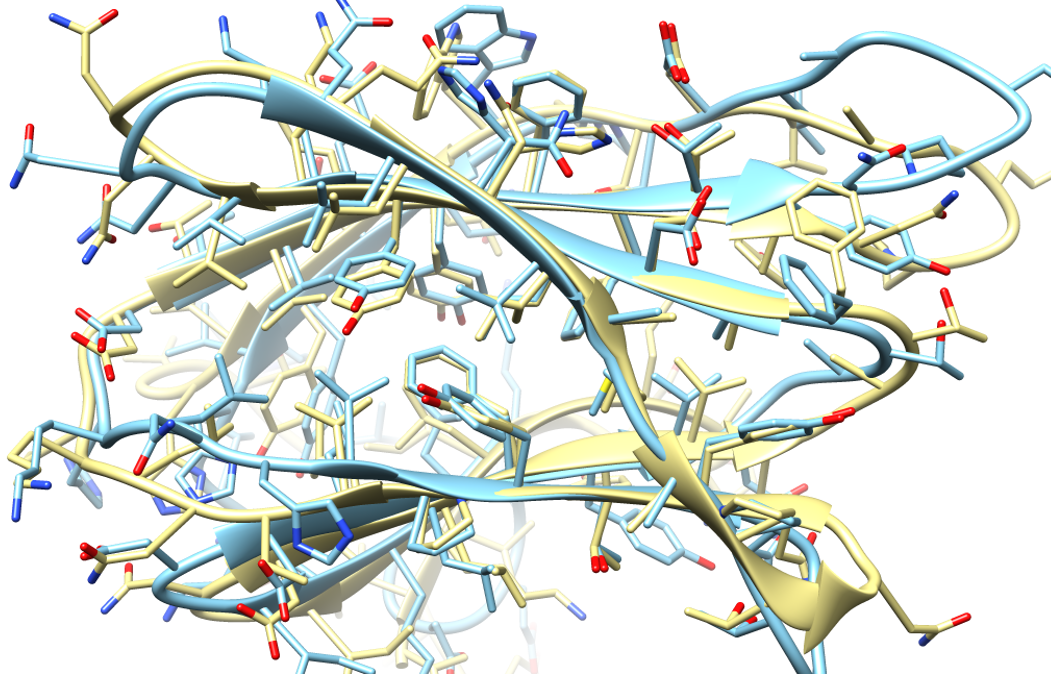
Protein structure prediction has long been available as an alternative to experimental structure determination, especially via homology modeling based on templates from related sequences. Recently, models based on distance restraints from coevolutionary analysis via machine learning to have significantly expanded the ability to predict structures for sequences without templates. One such method, AlphaFold, also performs well on sequences where templates are available but without using such information directly. Here we show that combining machine‐learning based models from AlphaFold with state‐of‐the‐art physics‐based refinement via molecular dynamics simulations further improves predictions to outperform any other prediction method tested during the latest round of CASP. The resulting models have highly accurate global and local structures, including high accuracy at functionally important interface residues, and they are highly suitable as initial models for crystal structure determination via molecular replacement.