
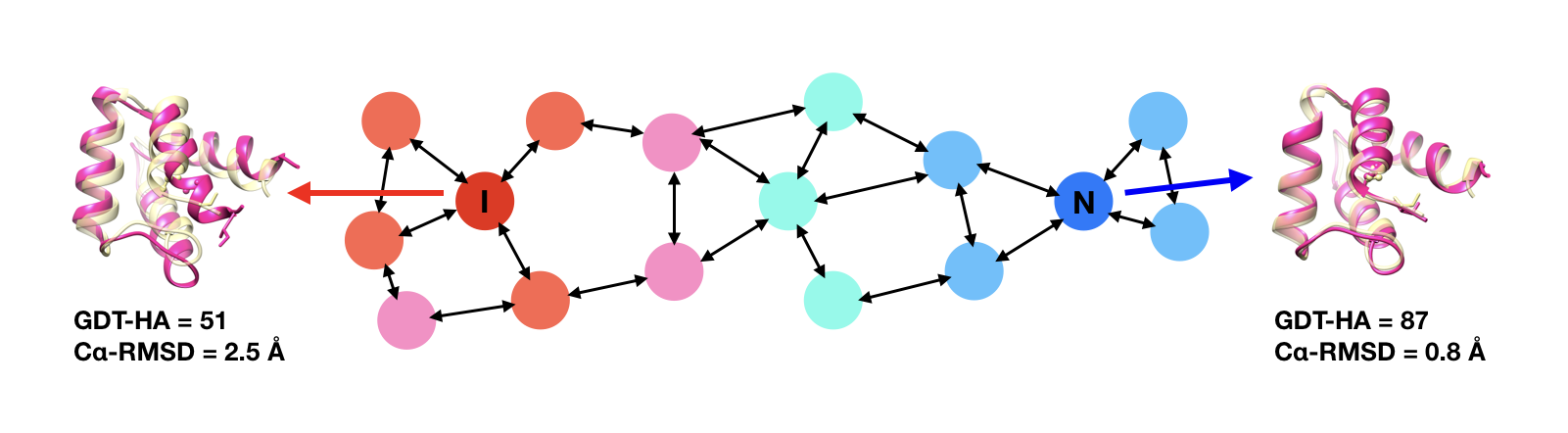
Computational protein structure prediction methods are widely used to generate models for gene sequences where protein structures are not available. Current methods perform very well, often generating models that are at least in terms of the overall fold correctly reproducing native structures. However, although close, most models do not reach experimental accuracy and therefore are not always useful for applications where highly accurate starting structures are required.
We are developing protein structure refinement methods with the goal of taking approximate models and bringing them closer to the native state. We have had success with molecular dynamics based protocols that involved iterative extensive sampling with latest generation force fields under limited restraints and ensemble averaging to more closely reflect how experimental structures are obtained. Our approach, PREFMD, performed very well during recent rounds of CASP by providing modest, but consistent refinement for almost all targets.
We also recently developed a second protocol, locPREFMD, that is aimed at local structure refinement where the backbone is preserved but MolProbity scores are greatly improved.
| 2020 | ||
| 17. | Lim Heo, Michael Feig: High-accuracy protein structures by combining machine-learning with physics-based refinement Proteins (2020) 88, 637-642 Abstract PDF | |
| 2019 | ||
| 16. | Lim Heo, Collin F. Arbour, Michael Feig: Driven to Near-Experimental Accuracy by Refinement via Molecular Dynamics Simulations Proteins (2019) 87, 1263-1275 Abstract PDF | |
| 2018 | ||
| 15. | Lim Heo, Michael Feig: Experimental Accuracy in Protein Structure Refinement via Molecular Dynamics Simulations Proceedings of the National Academy of Sciences, USA (2018) 115, 13276-13281 Abstract PDF | |
| 14. | Bercem Dutagaci, Lim Heo, Michael Feig: Structure Refinement of Membrane Proteins via Molecular Dynamics Simulations Proteins (2018) 86, 738-750 Abstract PDF | |
| 13. | Lim Heo, Michael Feig: PREFMD: A web server for protein structure refinement via molecular dynamics simulations Bioinformatics (2018) 34, 1063-1065 Abstract PDF | |
| 12. | Lim Heo, Michael Feig: What makes it difficult to refine protein models further via molecular dynamics simulations? Proteins (2018) 86, 177-188 Abstract PDF | |
| 2017 | ||
| 11. | Bercem Dutagaci, Michael Feig: Determination of Hydrophobic Lengths of Membrane Proteins with the HDGB Implicit Membrane Model Journal of Chemical Information and Modeling (2017) 57, 3032-3042 Abstract PDF | |
| 10. | Bercem Dutagaci, Kitiyaporn Wittayanarakul, Takaharu Mori, Michael Feig: Discrimination of Native-like States of Membrane Proteins with Implicit Membrane-Based Scoring Functions Journal of Chemical Theory and Computation (2017) 13, 3049-3059 Abstract PDF | |
| 9. | Michael Feig: Computational structure refinement: Almost there, yet still so far to go WIREs Computational Molecular Science (2017) 7, e1307 Abstract PDF | |
| 2016 | ||
| 8. | Michael Feig: Local protein structure refinement via molecular dynamics simulation with locPREFMD Journal of Chemical Information and Modeling (2016) 56, 1304-1312 Abstract PDF | |
| 7. | Michael Feig, Vahid Mirjalili: Protein structure refinement via molecular dynamics simulations: What works and what does not? Proteins (2016) 84 (Suppl 1), 282-292 Abstract PDF | |
| 2014 | ||
| 6. | Vahid Mirjalili, Kennan Noyes, Michael Feig: Physics Based Protein Structure Refinement through Multiple Molecular Dynamics Trajectories and Structure Averaging Proteins (2014) 82 (Supplement S2), 196-207 Abstract PDF | |
| 2013 | ||
| 5. | Vahid Mirjalili, Michael Feig: Protein structure refinement through structure selection and averaging from molecular dynamics ensembles Journal of Chemical Theory and Computation (2013) 9, 1294-1303 Abstract PDF | |
| 2010 | ||
| 4. | Michael Feig, Srinivasa Gopal, Kangasabai Vadivel, Andrew Stumpff-Kane: Conformational sampling in structure prediction and refinement with atomistic and coarse-grained models in: Multiscale approaches to protein modeling: Structure prediction, dynamics, thermodynamics and macromolecular assemblies, ed. A. Kolinski, Springer (2010) , | |
| 2008 | ||
| 3. | Mark A. Olson, Michael Feig, Charles L. Brooks III: Prediction of Protein Loop Conformations Using Multiscale Modeling Methods with Physical Energy Scoring Functions Journal of Computational Chemistry (2008) 29, 820-831 Abstract PDF | |
| 2. | Andrew Stumpff-Kane, Katarzyna Maksimiak, Michael S. Lee, Michael Feig: Sampling of near-native protein conformations during protein structure refinement using a coarse-grained model, normal modes, and molecular dynamics simulations Proteins (2008) 70, 1345-1356 Abstract PDF | |
| 2006 | ||
| 1. | Andrew Stumpff-Kane, Michael Feig: A Correlation-Based Method for the Enhancement of Scoring Functions on Funnel-Shaped Energy Landscapes Proteins (2006) 63, 155-164 Abstract PDF |